
Research Directions
Microbial metabolism, metabolomics, and isotope tracing
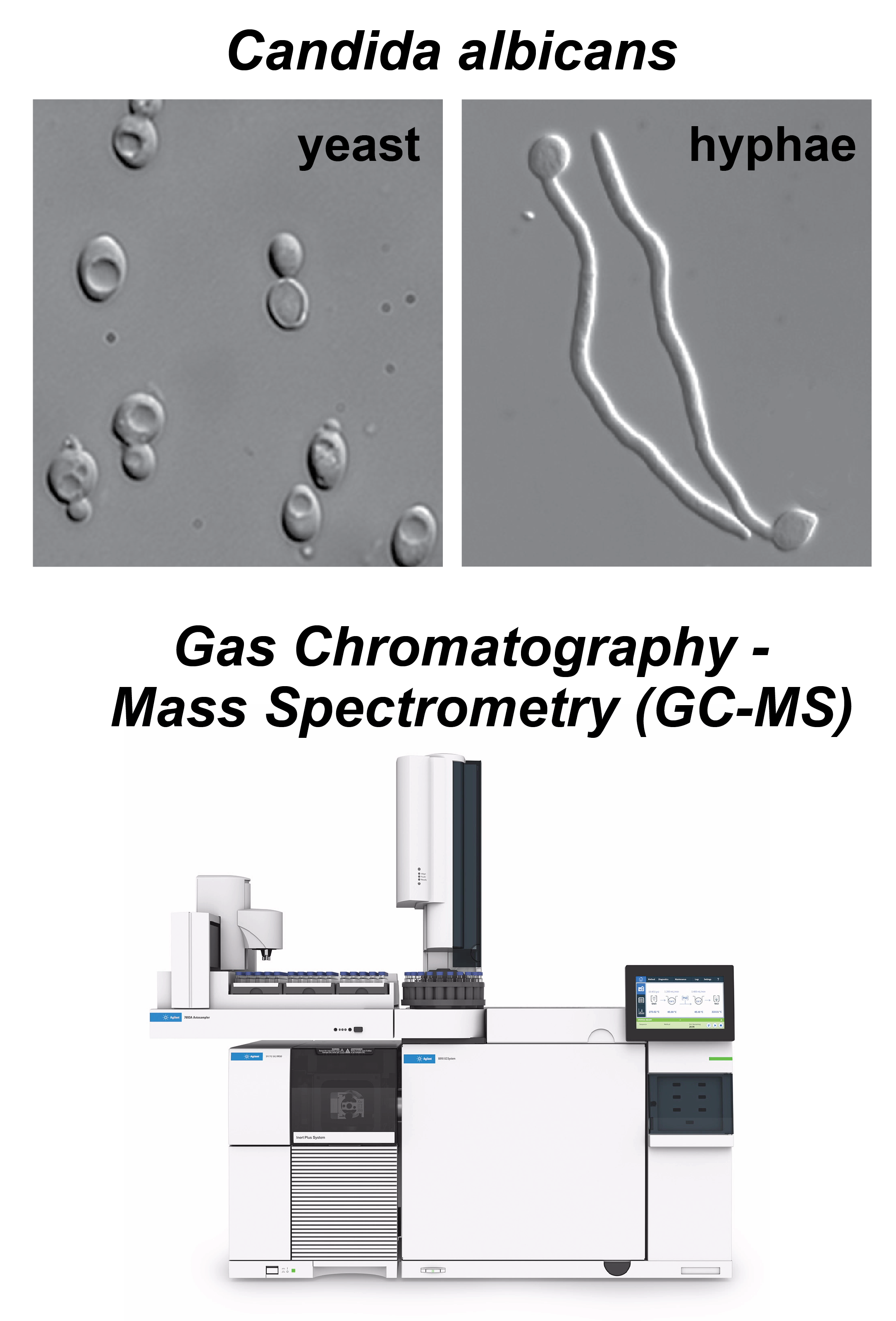 Adapted from Peter E. Sudbery, Nat Rev Microbiol (2011) . | We study microbial metabolism using metabolomics and isotope tracing to quantify metabolic fluxes across scales — from intracellular pathway activity to cross-feeding in multispecies communities. Our goal is to develop experimental and computational approaches that expand flux coverage while making flux quantification as accessible and routine as possible. A major focus of the lab is fungal pathogens. In Candida albicans, we investigate how metabolism is rewired during the transition between yeast and hyphal growth. In Aspergillus fumigatus, we study metabolic reprogramming during biofilm growth, with a particular emphasis on the spatiotemporal distribution of metabolites and fluxes. Beyond metabolomics, we are building a 3D biophysical modeling platform that integrates hyphal growth simulations with genome-scale metabolic modeling to infer spatially resolved metabolic activity within biofilm structures. Ultimately, we aim to link fungal metabolic states to clinically relevant phenotypes, including morphogenesis, biofilm architecture, antifungal resistance, and virulence, and to translate these insights into new metabolism-targeted antifungal therapies. |
Machine learning and AI for microbiology
 Generated by Gemini. | We develop machine learning and artificial intelligence (AI) approaches to predict microbial phenotypes from genomic information and host gastrointestinal phenotypes from gut microbial communities. A major focus is the development of species-specific genomic language models that learn microbial sequence context and regulatory logic to predict how genetic variation affects gene expression, fitness (e.g., growth), and function (e.g., antibiotic resistance). We also apply supervised and interpretable machine learning to multi-omics datasets from cystic fibrosis (CF) cohorts to predict gut/lung inflammation from microbiome and identify microbial/host pathways associated with clinical treatment (e.g., CFTR modulator) outcome heterogeneity. By integrating "black-box" machine learning models with "white-box" mechanistic approaches, we aim to identify multi-scale molecular signatures as biomarkers that explain variation in microbial and host phenotypes in patients with CF. Ultimately, our goal is to build modern AI tools that enable multi-omics-informed diagnostics and personalized therapeutic strategies. |
Gut microbiome ecology and host-microbe interactions
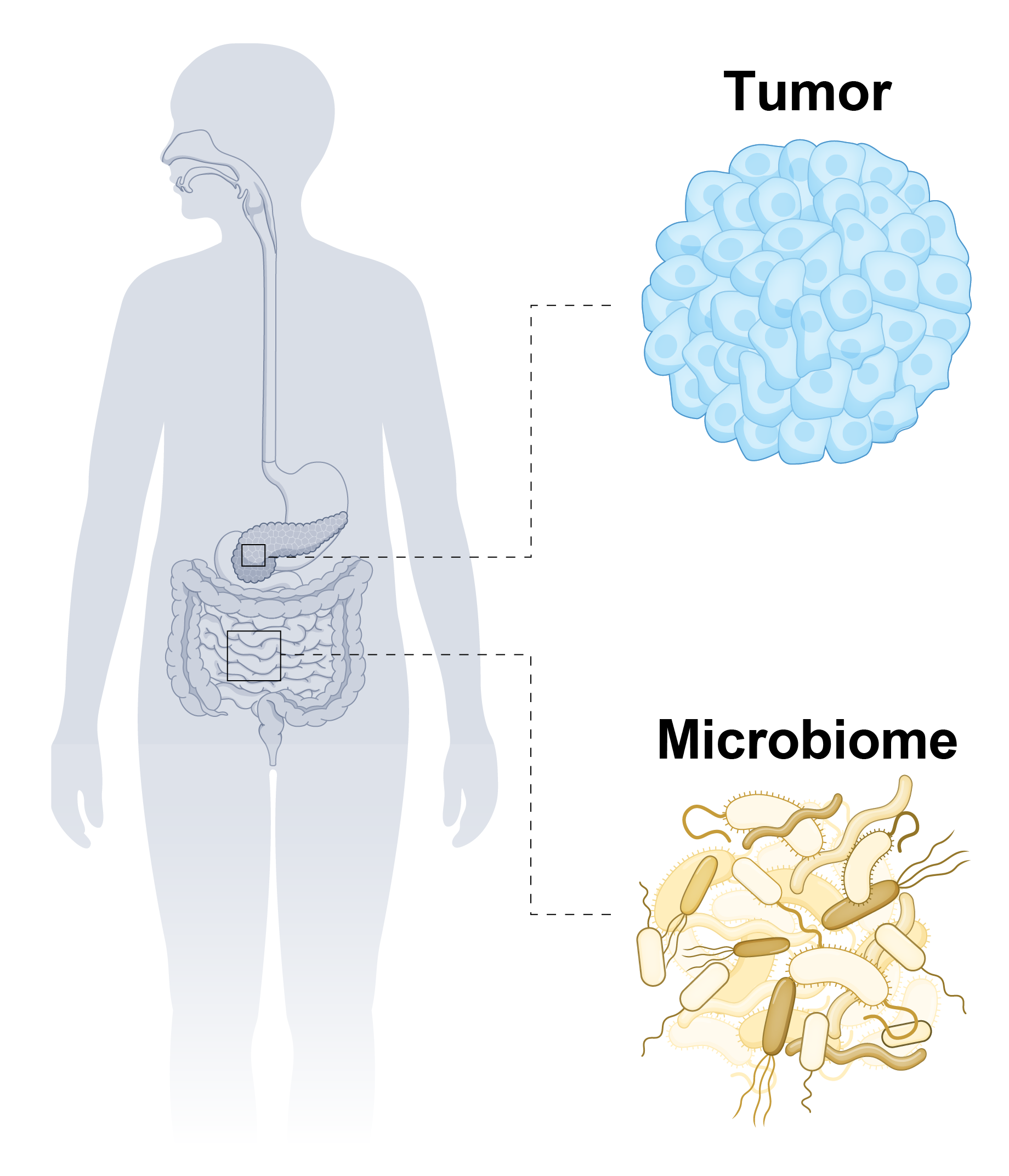 Adapted from Ryan M. Thomas, Nat Rev Gastroenterol Hepatol (2025) . | We develop experimental and computational approaches to understand how microbiome composition and function shape host immunity and modulate responses to cancer immunotherapy. Our data show that gut colonization by Bacteroides spp. reduces colorectal tumor growth in mice, motivating mechanistic questions about how their colonization alters host immune programs and metabolism. We study these interactions using complementary in vitro, in vivo, and in silico models. We use co-culture platforms and isotope tracing to track metabolite flow between Bacteroides spp. and tumor cells or host immune cells, and we collect single-cell rRNA sequencing, spatial transcriptomics, and spatial metabolomics data from mouse tissues to quantify host responses in the tumor microenvironment. These measurements are integrated with statistical and mechanistic modeling, including models of immune cell population dynamics and microbiome–immune regulation, to identify microbes, metabolites, and metabolic pathways that enhance or suppress anti-tumor immunity. Ultimately, we aim to identify gut-derived molecules that regulate host responses to immunotherapy and use these insights to faciliate rational design of microbiome-based therapies. |